Nach 25 Jahren als Leiter der DOG-Sektion Kornea verabschiedet sich Herr Prof. Berthold Seitz aus dem Vorsitz
Weiterlesen 1693
1693
 1693
1693
 580
580



 1303
1303

 607
607
 1241
1241
 1421
1421



 601
601

 550
550


 566
566




 874
874
 737
737

 1808
1808
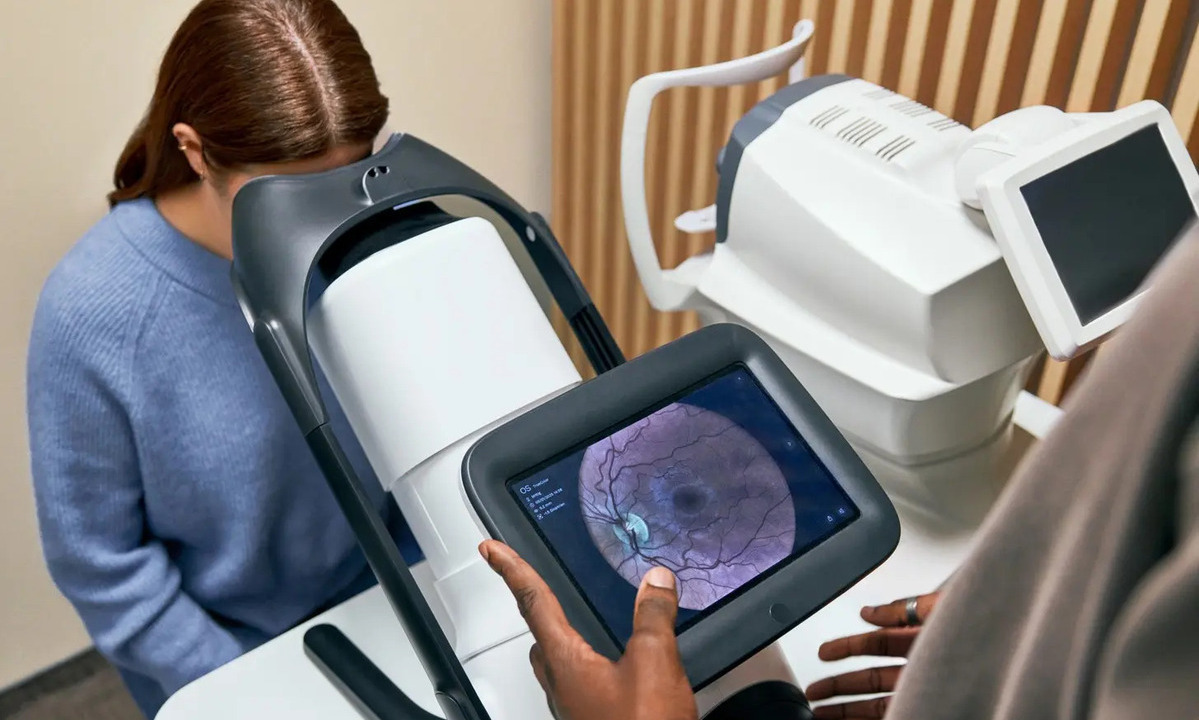
 946
946
 1130
1130
 2010
2010
 1005
1005

 2563
2563

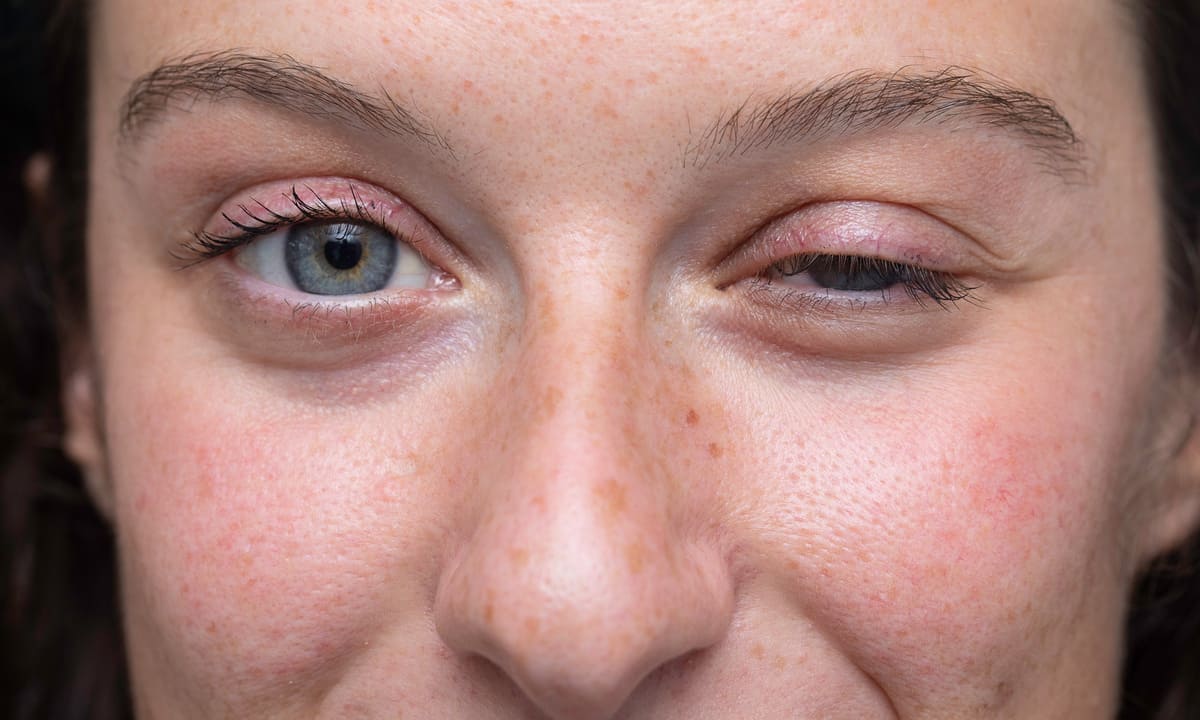
 816
816

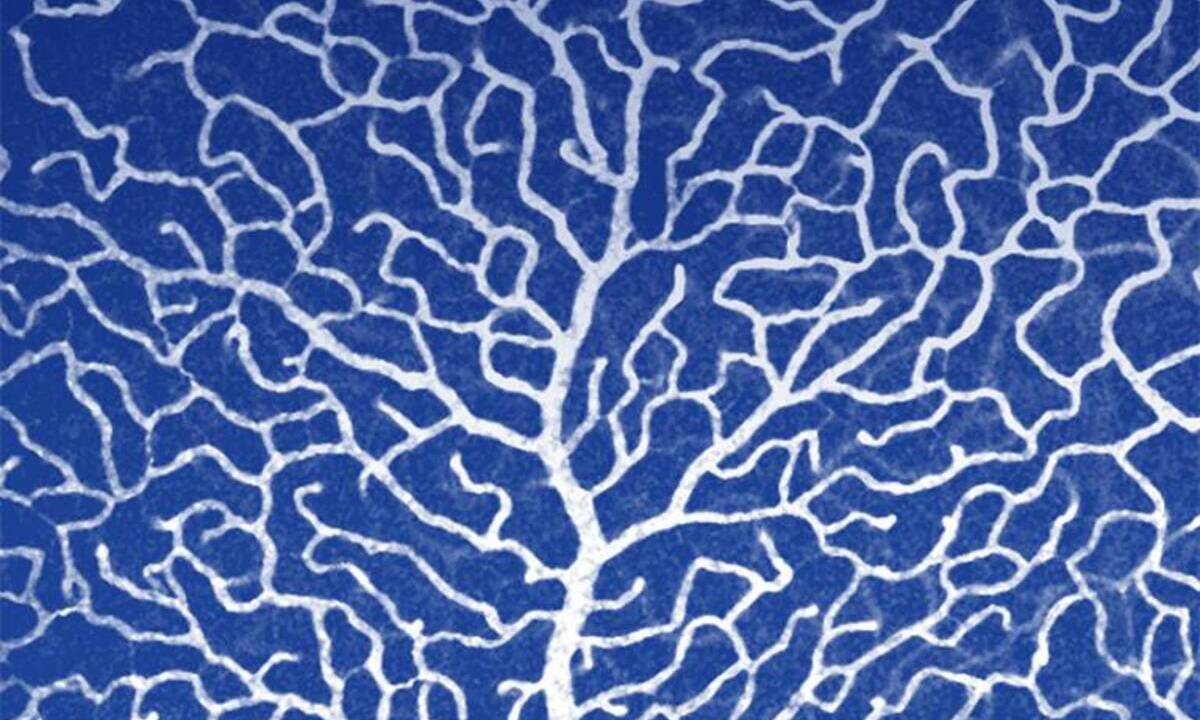 3989
3989
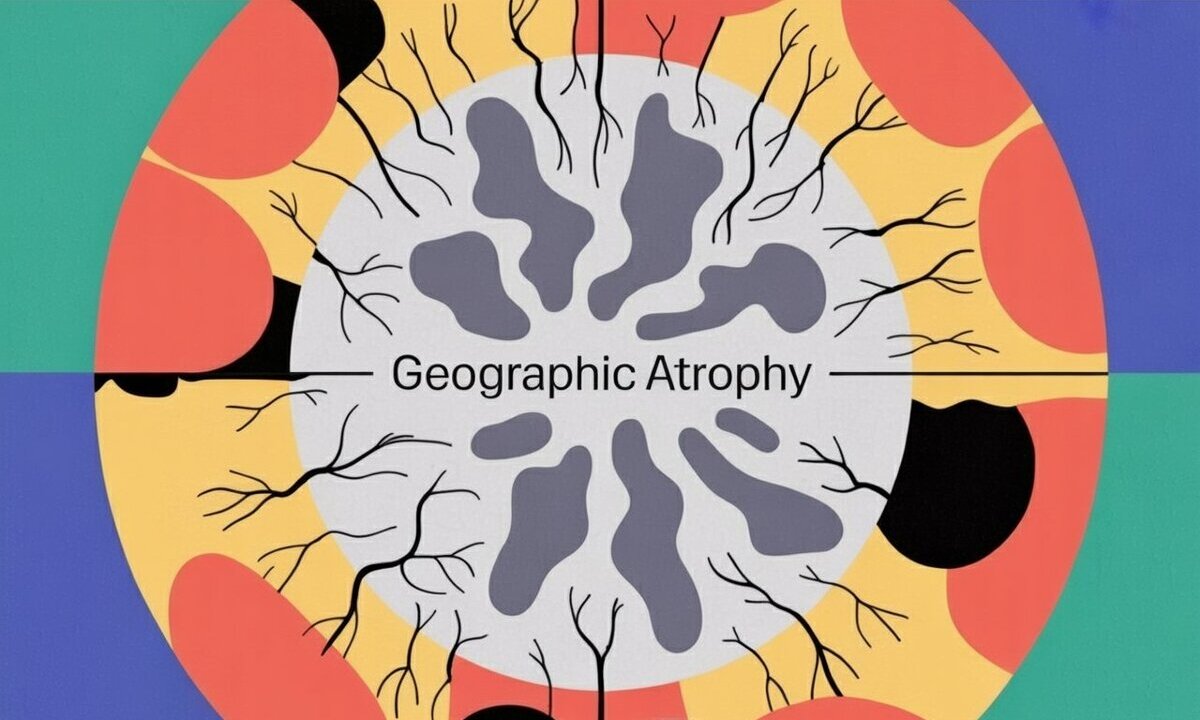 1790
1790

 2149
2149
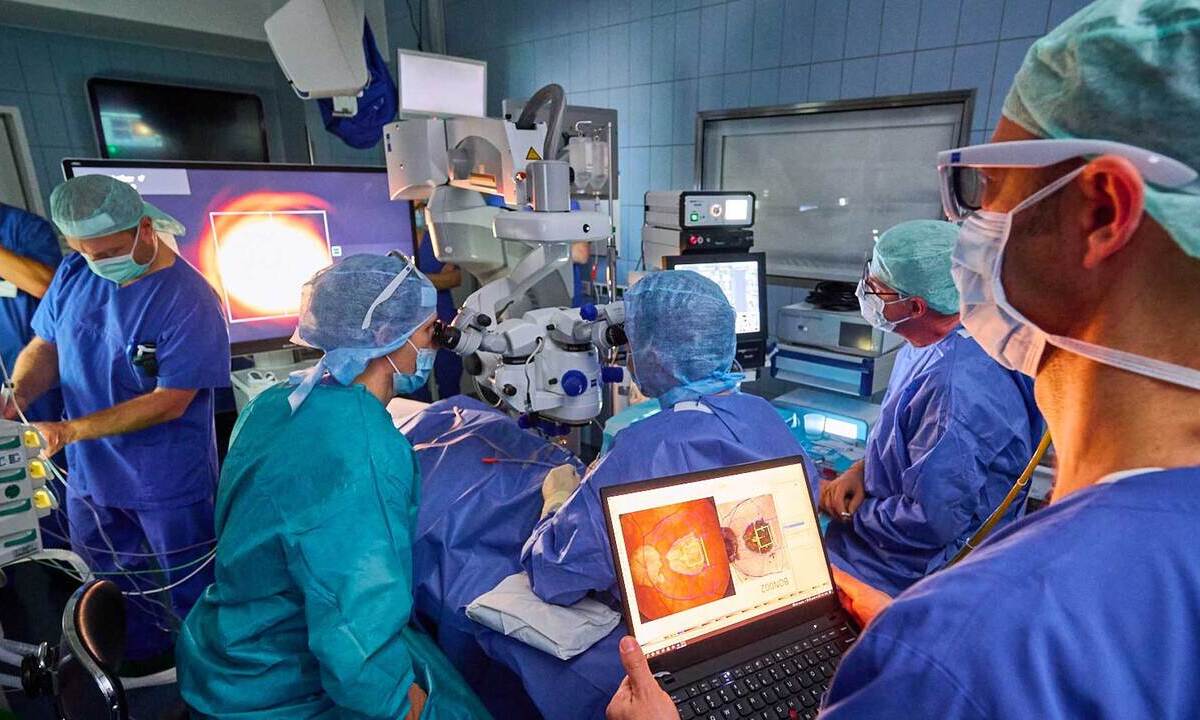





 1229
1229


 1495
1495
 630
630

 1415
1415
 799
799

 1145
1145
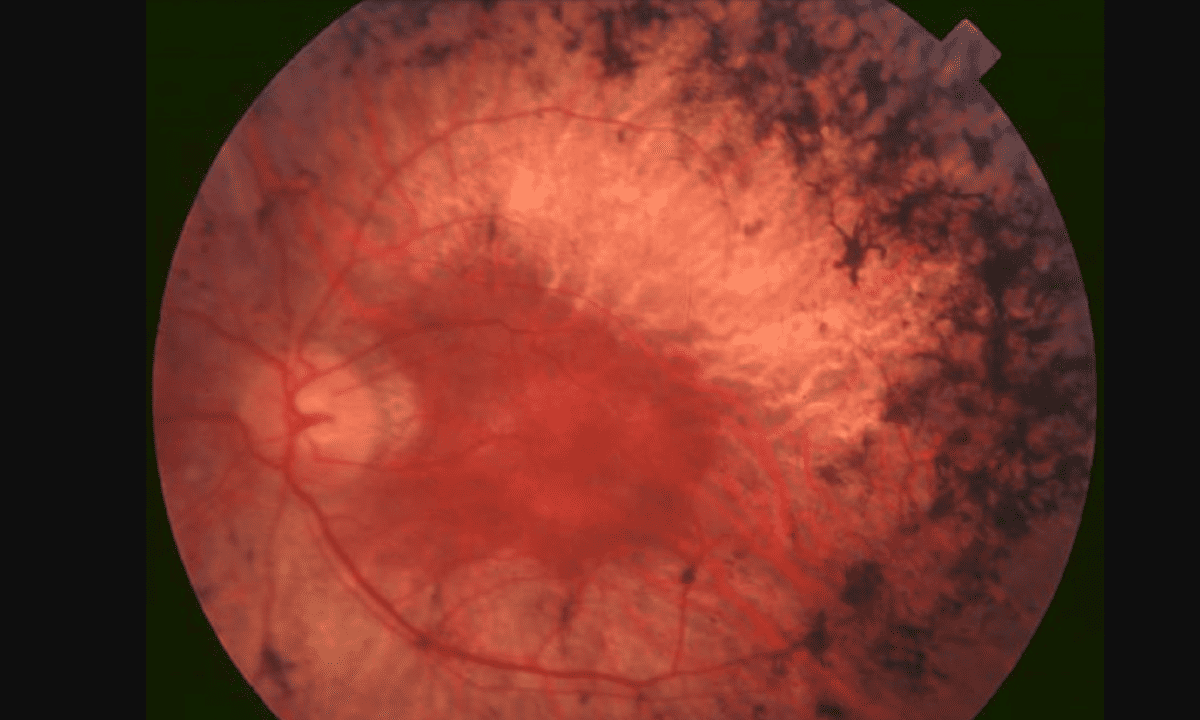
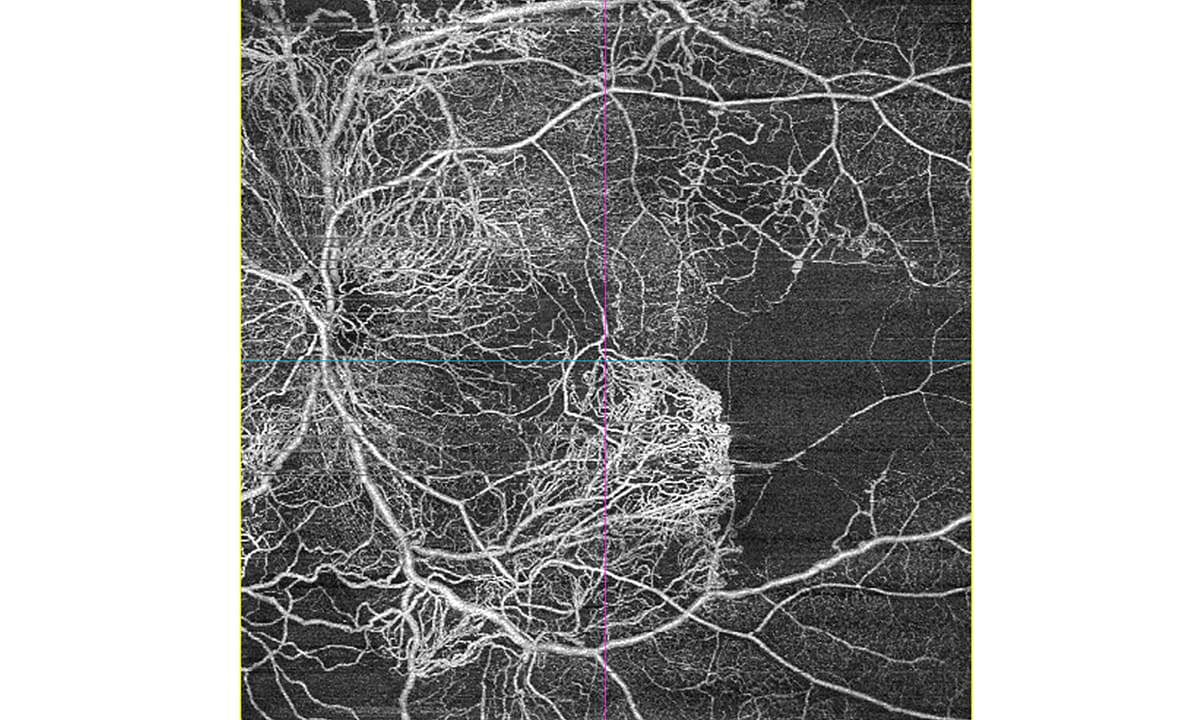 5544
5544
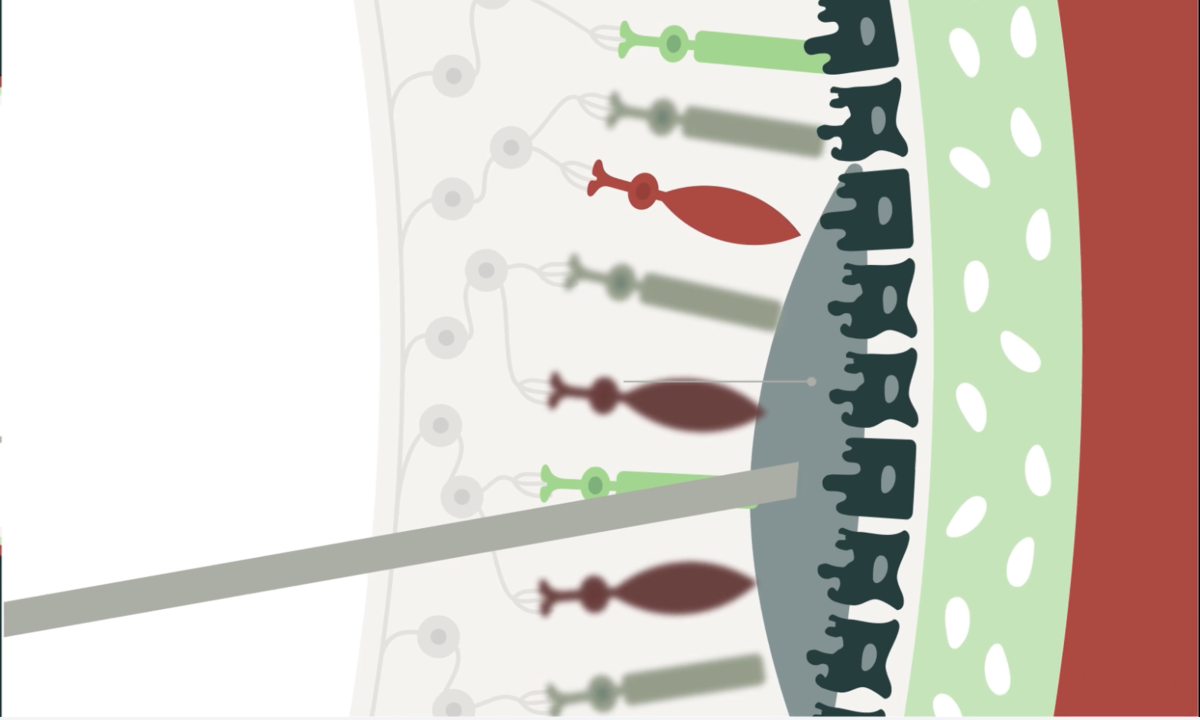 725
725

 1274
1274
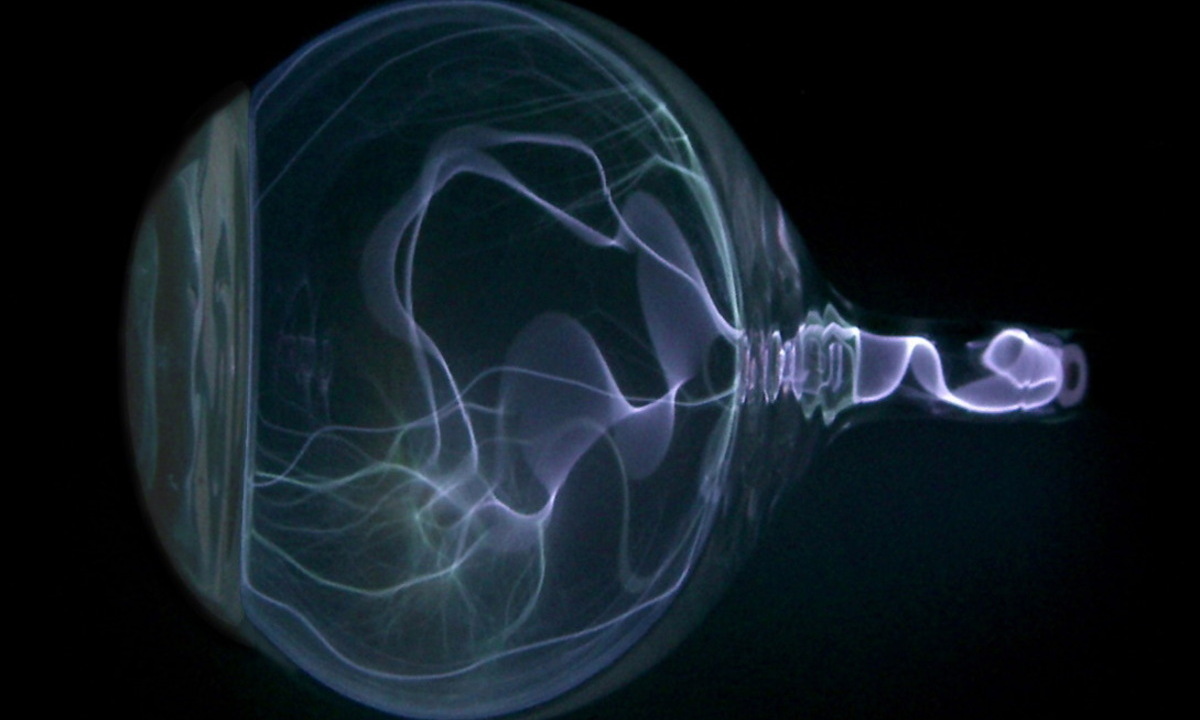


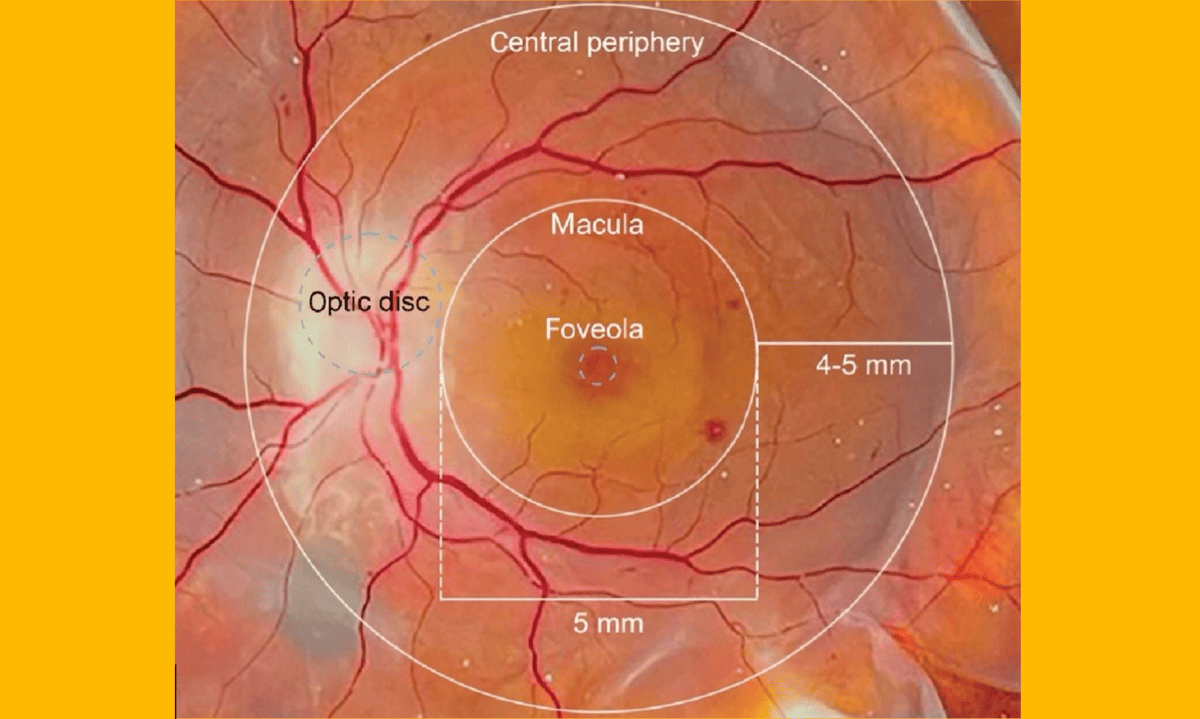



 975
975


 1621
1621
 1857
1857
 949
949

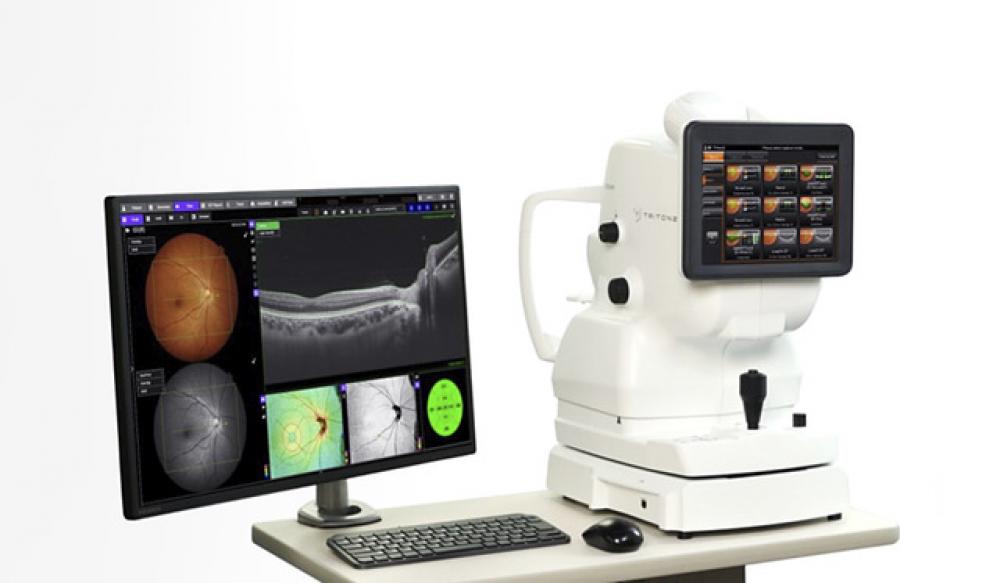 723
723
 771
771


Fluocinolonacetonid ist EINFACH, VERLÄSSLICH und WIRKSAM
Weiterlesen 1260

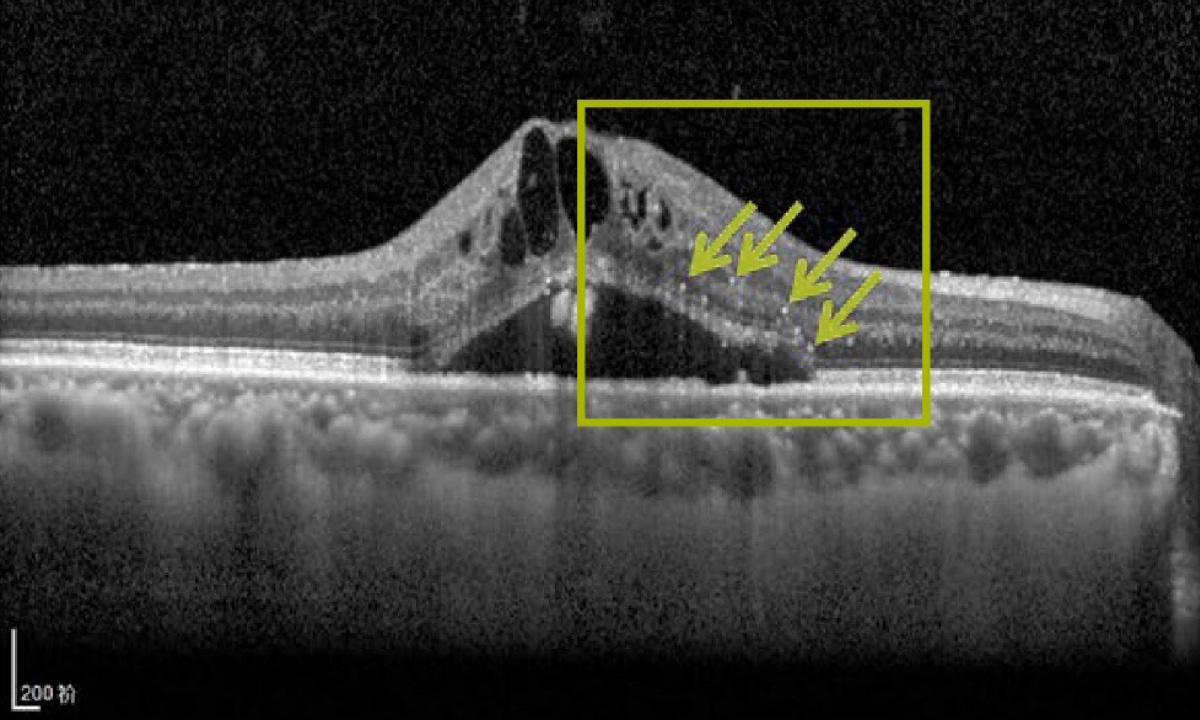 1205
1205
 1062
1062

 2712
2712

 1847
2095
1847
2095


Eyefox Mitglieder erhalten Rabatt