Der blinde Fleck des Genoms - Forschende vom IOB decken Ursache für erblichen Sehverlust auf
Obwohl mehr als hundert Gene mit Retinitis pigmentosa (RPP) in Verbindung gebracht worden sind, bleibt die genetische Ursache bei ~30-40% der Patienten ungeklärt, selbst nach umfangreichen DNA-Tests. Für betroffene Familien bedeutete dies eine jahrelange Unsicherheit über die Ursache ihrer erblichen Erkrankung. Doch diese Ungewissheit beginnt sich nun aufzulösen.
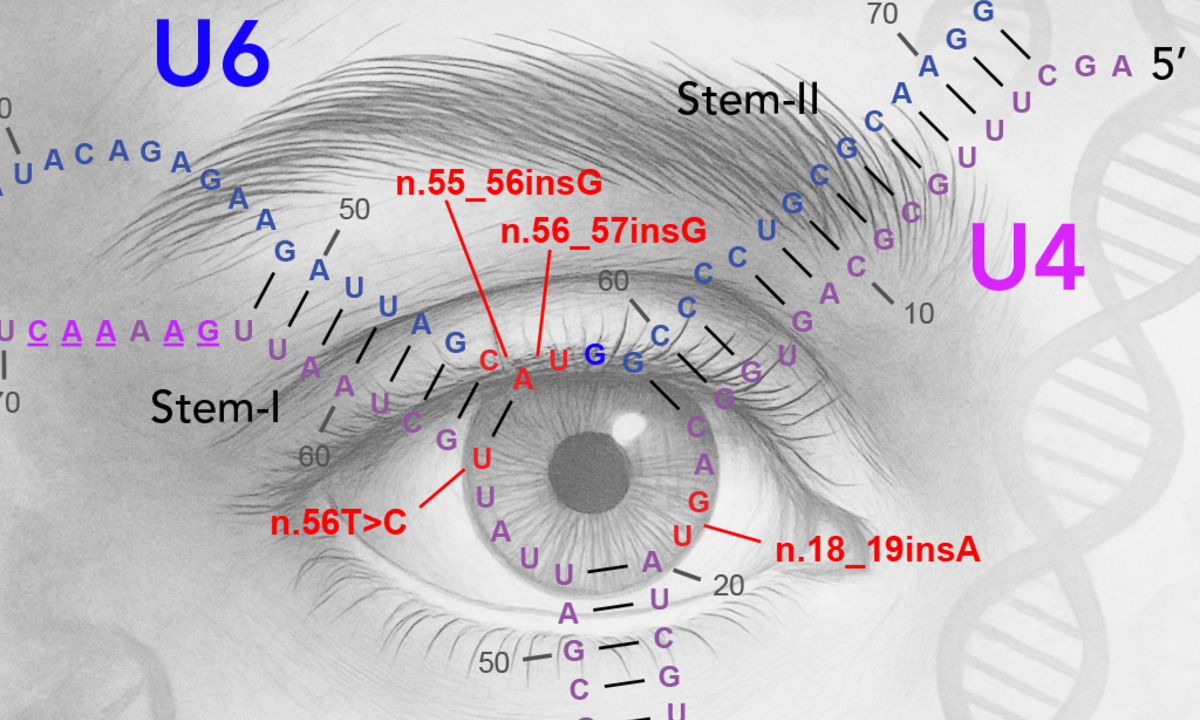
Forschende des Instituts für Molekulare und Klinische Ophthalmologie Basel (IOB) haben in Zusammenarbeit mit mehr als 100 Institutionen weltweit genetische Daten von fast 5'000 Personen aus 62 von RP betroffenen Familien analysiert. Die krankheitsverursachenden Mutationen wurden aber nicht in proteinkodierenden Genen gefunden. Bei 153 Patienten identifizierten die Forschenden stattdessen Veränderungen in RNA-Molekülen, die am Spleissmechanismus der Zelle beteiligt sind, welcher genetische Informationen bearbeitet, bevor Proteine hergestellt werden.
Die wichtigsten Ergebnisse in Kürze:
- Varianten in fünf nicht-kodierenden RNA-Genen (RNU4-2, RNU6-1, RNU6-2, RNU6-8 und RNU6-9) verursachen Retinitis pigmentosa. Diese Gene produzieren RNA-Moleküle anstelle von Proteinen und stellen eine weitgehend unerforschte Quelle vererbbarer Blindheit dar.
- Die Varianten sind sowohl vererbt als auch spontan. Einige wurden über Generationen weitergegeben, andere traten zum ersten Mal bei betroffenen Personen auf.
- Alle Varianten konzentrieren sich in derselben kritischen Region, wo sich die U4-und U6-RNA-Moleküle, die von den RNU4- und RNU6-Genen auf der DNA codiert werden, verbinden. Dies ist eine wichtige Interaktionsstelle für mehrere am RNA-Spleissen beteiligte Proteine.
- Ein und dasselbe Gen kann verschiedene Krankheiten verursachen. Während bestimmte Varianten in RNU4-2 zu neurologischen Entwicklungsstörungen führen, zielen die hier identifizierten Varianten spezifisch auf die Netzhaut ab.
Diese Entdeckung schliesst eine wichtige Lücke. Dass Mutationen in bestimmten Spleiss-Proteinen (PRPF3, PRPF8 und PRPF31) RP verursachen, war bereits bekannt. Diese Studie zeigt nun, dass auch die RNA-Moleküle des Spleissmechanismus krankheitsverursachende Varianten aufweisen können. Mit anderen Worten: Verschiedene Komponenten desselben zellulären Prozesses führen, wenn sie defekt sind, zur selben Erkrankung.
Diese Varianten erklären bis zu 1,4% der bisher nicht diagnostizierten RP-Fälle. Dies bedeutet, dass Dutzende von Familien weltweit nun eine präzise molekulare Diagnose erhalten können. Sie können genetische Beratung in Anspruch nehmen, informierte Entscheidungen über Familienplanung treffen und sich für zukünftige Behandlungen positionieren, sobald sie verfügbar sind.
Die Studie vertieft ausserdem das Verständnis der Ursachen erblicher Blindheit massgeblich. Der Blick in bisher übersehene, nicht-proteinkodierende Regionen des Genoms eröffnet neue diagnostische Wege. Während sich genetische Tests weiterentwickeln und RNA-basierte Therapien voranschreiten, legen diese Erkenntnisse wesentliche Grundlagen für die Identifizierung weiterer Patienten und letztendlich für die Entwicklung von Behandlungen für eine Krankheit, die derzeit noch unheilbar ist.
Der vollständige Artikel “De novo and inherited dominant variants in U4 and U6 snRNA genes cause retinitis pigmentosa” ist erschienen in Nature Genetics und hier abrufbar.
Quelle: IOB